
骆智训课题组在簇合物电子规则与化学键理论研究方面取得新进展
化学键理论是研究原子间相互作用及分子结构的基础,旨在解释化学键的本质、稳定性和分子几何构型。随着量子力学的发展,化学键理论逐步完善。从早期的经典价键理论和分子轨道理论,到现代广泛应用的密度泛函理论,其研究范畴不断拓展,并广泛应用于材料科学、催化化学和生物分子模拟等领域。化学键理论的发展不仅推动了对分子世界的理解,还为新材料的理性设计提供了依据。近年来,簇合物的合成与化学研究蓬勃发展,但多核金属簇的成键机制仍不完全清楚,传统理论难以精确描述其电子结构。
在国家自然科学基金委、中国科学院及北京市自然科学基金委的支持下,化学所分子动态与稳态结构实验室骆智训课题组基于自主研制的团簇科学实验装置,长期致力于金属团簇的精准制备及其化学反应研究,广泛探索了活性与惰性金属团簇的反应性与相对稳定性机制(J. Phys. Chem. Lett. 2025,16,454−459; 2025,5538−5545; 2024,15,11383−11394; 2024,15,1856−1865; 2024,15,9888−9893; 2022,13,9711−9717; 2022,13,9711−9717; 2022,13,733−739; 2021,12,5115−5122; 2021,12,1593−1600; 2020,11,8222−8230; 2020,11,5807−5814; 2012,3,3818−3821),并基于狭义和广义超原子概念(Acc. Chem. Res. 2014,47,2931–2940; Coord. Chem. Rev. 2024,500,215505)深入诠释了金属团簇中的电子离域现象和多中心键规律。
近期,该课题组和合作者通过Rhn±和Ptn±团簇与NO、CO分子的反应,首次成功制备了一系列高选择性金属羰基及亚硝酰基配合物。研究发现,高选择性的Ptn(NO)n±和Ptn(NO)n-1±团簇复合物均符合xe@12e电子构型,即每个Pt原子局域配位12电子,而离域电子则呈现自适应平衡的规律。基于此,研究团队针对多核金属簇合物提出了一种新的电子计数规则,阐明了金属团簇稳定化过程中离域电子与局域配位饱和趋势的动态平衡机制,将其简称为“金属团簇电子离域与平均配位饱和自适应平衡规则(electronic"Aā"rule)。该理论能够合理解释已知多核簇合物的稳定性机制,并成功预测了Pt8(NO)7‒等团簇的核壳结构与电子构型,与光电子谱研究结果一致。
该项工作通过对一系列高选择性团簇配合物的深入研究,揭示了离域电子和局域电子在成键过程中的协同作用机制,为簇合物的稳定性与结构预测以及新材料设计提供了新的见解和思路。相关成果发表在Chem. Sci. 期刊上(论文链接:10.1039/D5SC02924D)。论文的共同第一作者为化学所博士生林世泉和安徽大学李丹,通讯作者是安徽大学程龙玖教授和化学所骆智训研究员。
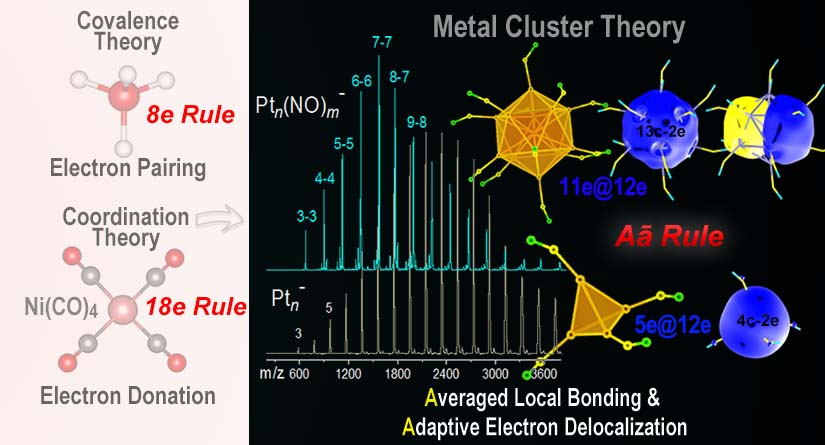
图1. 从八电子规则到十八电子规则再到金属团簇电子计数规则(电子离域与平均配位饱和自适应平衡规则);
图中质谱显示纯金属Ptn−团簇正态分布及其与NO反应后形成高选择性团簇配合物。
分子动态与稳态结构实验室
2025年6月4日
附件下载:


